


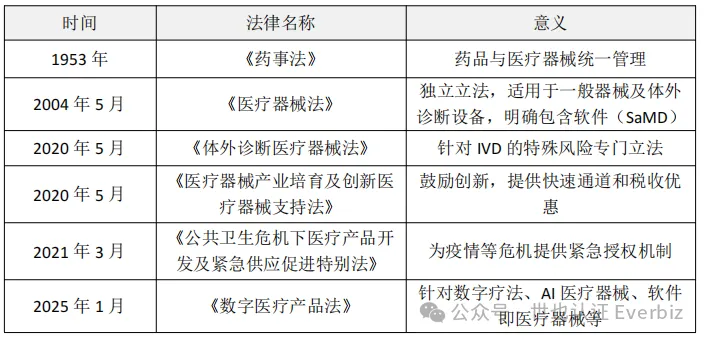
韩国医疗器械监管法律经历了从“药械合一”到专门立法的演变过程:
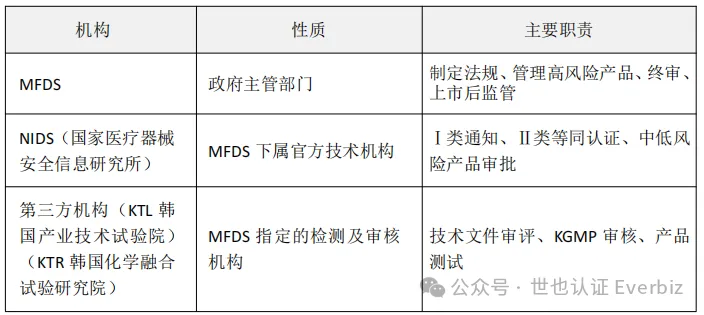
实际影响:
l一般有源/无源器械(ClassⅠ–Ⅳ)→适用《医疗器械法》 l体外诊断试剂/设备(ClassⅠ–Ⅳ)→适用《体外诊断医疗器械法》 l创新医疗器械→可申请“创新医疗器械指定”获得加速 l数字疗法、AI诊断软件→2025年起适用《数字医疗产品法》


Class I(低风险,如口罩、体温计)
Class II(中风险,如超声、监护仪)
Class III / IV(高/最高风险,如支架起搏器)

韩国对实质等同的II类器械采用高效两阶段流程:
阶段1:第三方机构技术文件评审(KTL/KTR等)
提交评审申请:技术文件包括产品性能报告、实质等同性比对数据、风险管理报告、生物相容性/电气安全测试报告等。 评审+补正:第三方全面审查,必要时要求补充资料。 签发《符合性函》:承诺25天内签发(不含补正时间)。
阶段2:NIDS认证发证
提交认证申请:附上《符合性函》及行政资料(韩文标签、说明书、韩国授权代理商信息等)。 行政评审+补正:NIDS仅做形式审查,不重复技术审查。 签发注册证书:承诺5天内完成(不含补正时间)

Review Request:向MFDS提交技术文档审评请求。 Application:通过NIDS系统提交正式申请。 Review:MFDS审评, 可能要求补充资料。

分类与指定规定 批准/认证/通知/评价规定 数字医疗器械GMP(QMS) 临床试验方案审批与实施管理 网络安全规定 卓越治理体系认证标准
l数字医疗器械软件授权审评指南(新设) l应用AI技术的数字医疗器械临床试验设计方法指南(修订) l医疗器械软件授权审评指南(修订) l应用AI技术的数字医疗器械授权审评指南(修订) l应用虚拟融合技术的数字医疗器械授权审评指南(修订) l数字疗法授权审评指南(修订)

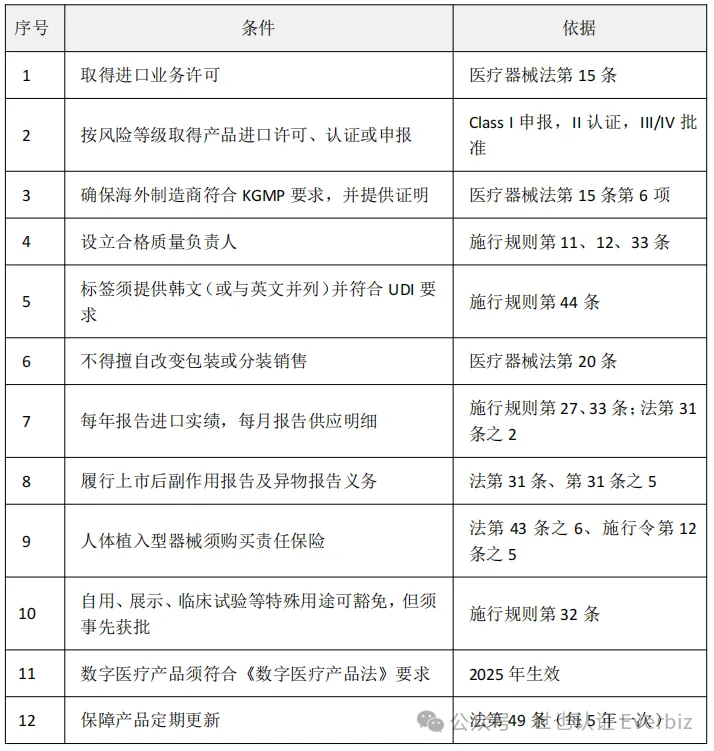
给中国企业的实操建议
提前确定产品分类:对照MFDS《医疗器械品目及品目别等级规定》,确认风险等级。 选择合适路径
有等同产品→ 走NIDS认证(2–4个月)
创新/数字产品 → 准备临床/算法资料,走MFDS许可(4–12个月)
加强上市后监测:植入式医疗器械长期随访要求,生效日期:2025年1月31日。 KGMP与MDSAP联合审核,2025年4月7日。变化:原KGMP和MDSAP分别审核,现面向出口制造商实施联合审核,减轻审计负担。 敏感信息处理及真实世界数据(RWD)收集。生效日期:2025年8月1日。内容:为上市后监测提供法律依据,允许收集和分析真实世界数据。
韩国医疗器械市场准入门槛清晰、监管成熟,是值得长期布局的优质海外市场。对于计划拓展全球器械版图的企业而言,提前掌握 MFDS 注册、KGMP 体系、数字医疗合规等核心要点,能为未来出海做好充分准备。
EVERBIZ 目前已深耕越南、新加坡、泰国、马来西亚、德国、奥地利、等主流市场,专注为医疗器械企业提供一站式海外注册、合规辅导与市场准入服务,凭借成熟的本地化资源与专业经验,帮助众多中国器械产品顺利进入海外市场。



