
日本是全球第二大医疗器械市场,也是中国医疗器械企业出海的重要目标市场。其法规体系以严谨、细致著称。全文基于日本厚生劳动省(MHLW)官方解读及行业实操指南,为您一站式梳理日本医疗器械注册的全貌,涵盖法律架构、风险分类、上市路径、核心标准、实施经验,助力您高效合规进入日本市场。

一、日本医疗器械法规体系概览
日本医疗器械的主要监管法律是:《药品和医疗器械法》(PMD Act),该法于2014年11月由原《药事法》修订更名而来,与以下法规共同构成完整监管体系:

监管机构
1️⃣ 厚生劳动省(MHLW):负责法规制定与最终批准。
2️⃣ 药品与医疗器械管理局(PMDA):负责技术审评、上市后监管、不良事件监测、反应救济补偿
二、日本医疗器械分类体系
日本采用基于风险的分类管理,共分为四类:
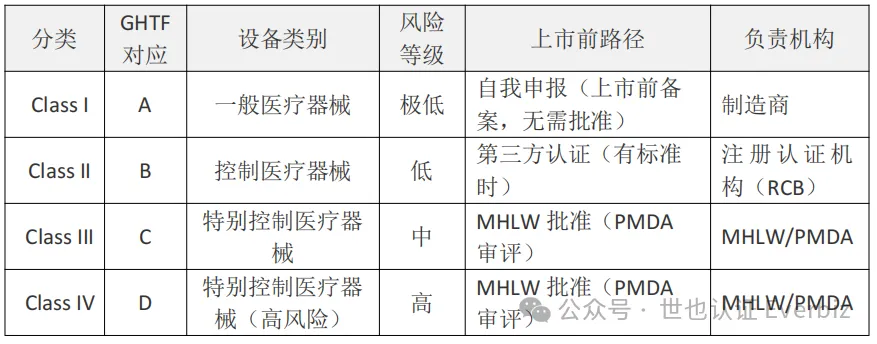
日本分类体系与GHTF风险分类体系对应:
Class I → A类
Class II → B类
Class III → C类
Class IV → D类
三、JMDN编码体系
日本采用:JMDN(Japanese Medical Device Nomenclature)
该体系基于全球GMDN建立。
编码结构:前5位:GMDN主编码,后3位:日本细分编码
其中:第6位反映风险等级,后2位用于产品细分
✅ 企业在注册前,必须首先确认JMDN代码。
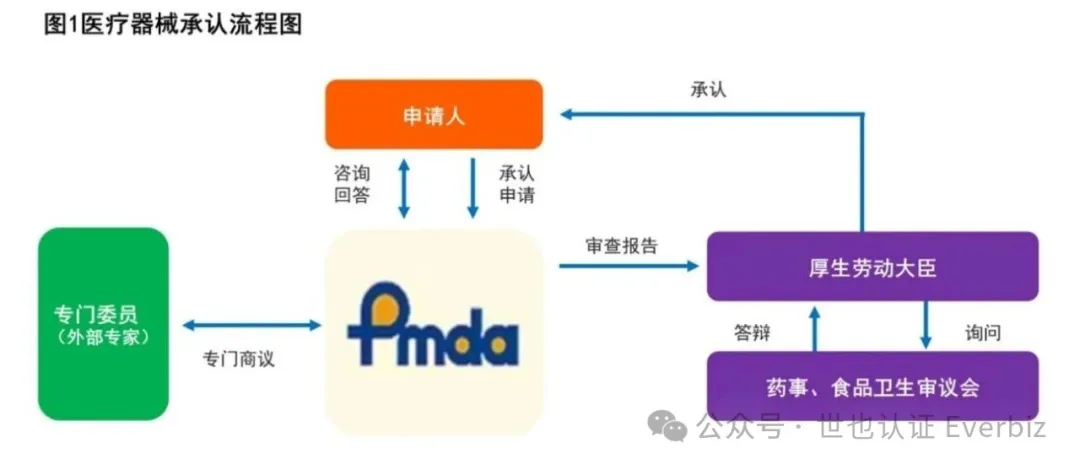
四、日本医疗器械上市路径
1️⃣ Class I —— 上市前备案(届出)
特点:无需PMDA技术审评,无需批准,企业自行确认符合基本原则
适用于:低风险器械,一般耗材
2️⃣ 上市认证(認証)
适用于:有认证标准的Class II,部分Class III产品
由谁审核?由:注册认证机构(RCB)进行审核,类似欧盟公告机构(NB)。
常见提交:资料产品规格、风险分析、性能测试、基本原则符合性、标准符合性声明
3️⃣ 上市批准(承認)
适用于:无认证标准的Class II、Class III、Class IV
创新产品需要提交:完整技术文档、临床评价、稳定性数据、风险管理文件
制造信息PMDA分类逻辑,PMDA会将产品区分为:新器械、改良器械、仿制器械、不同路径对临床要求差异很大。
五、日本认证标准体系
1️⃣ Certification Criteria(认证标准)
主要适用于:仿制器械、实质等同产品
特点:可第三方认证、引用JIS/ISO标准、需提交基本原则符合性
2️⃣ Approval Criteria(批准标准)
适用于:无需临床数据的产品
特点:技术要求统一、审批一致性高
3️⃣ Review Guidelines(审评指南)
用于:安全有效性评估、PMDA技术审查参考
特别适用于:创新器械、改良型器械
六、日本最新法规修正重点
近年来,日本大力推动创新器械快速上市。
1️⃣ SAKIGAKE先驱审查制度
适用于:全球同步开发产品、临床优势明显产品
优势:✅ 优先咨询✅ 专人审评✅ 缩短审批时间
2️⃣ 特定用途优先审评
针对:儿科器械、AMR抗耐药相关器械、高未满足需求产品
3️⃣ 有条件批准制度
特点:允许上市后继续补充数据、加快创新产品上市
4️⃣ PACMP上市后变更管理方案
企业可提前提交:规格变更、制造商变更、零部件变更、获批后无需每次重新申请。
✅ 大幅提升产品迭代效率。
七、外国企业进入日本的核心要求
1️⃣ 必须指定MAH
MAH(Marketing Authorization Holder)
即:日本上市许可持有人,MAH必须是日本本土法人。
2️⃣ 境外生产场所注册
需要注册的环节包括:设计开发、生产、灭菌、灌装
3️⃣ 建立日本QMS体系
日本QMS基于:ISO 13485,但要求更严格。
尤其重视:设计控制、风险管理生命周期管理
4️⃣ 接受现场审核
日本认可:MDSAP证书、可替代部分PMDA现场审核。
八、日本注册资料要求
常见资料包括:
✅ 产品规格
✅ 风险分析(ISO14971)
✅ 性能测试
✅ 稳定性研究
✅ 临床评价
✅ 灭菌验证
✅ 基本原则符合性
✅ 生产工艺文件
⚠️ 所有提交文件必须为:日文
九、日本注册周期与费用

十、日本上市后监管
日本采用三大安全体系:
1️⃣ 安全性审评
2️⃣ 上市后监测
3️⃣ 不良反应救济
不良事件上报方式企业可通过:电子提交、邮寄、现场递交
PMDA会进行:数据分析、因果关系评估、风险趋势研判
近年来,日本大量采用:
✅ 数据挖掘
✅ 哨点医院网络
✅ 主动风险预警
十一、企业实操建议
✅ 1. 提前确认分类
重点明确:是否实质等同、是否属于创新器械
✅ 2. 尽早选择MAH
MAH质量直接影响注册效率。
✅ 3. 充分利用快速通道
例如:SAKIGAKE、特定用途优先审评、有条件批准
✅ 4. 提前建立QMS体系
建议:ISO13485、MDSAP、同步推进。
✅ 5. 寻求专业团队支持
日本法规:文件要求严格、日文要求高、审评逻辑细致、专业团队可大幅提高成功率。

十二、常见问题 Q&A
Q1:日本医疗器械法规核心是什么?
A:PMD Act(《药品和医疗器械法》)。
Q2:Class I需要批准吗?
A:不需要,仅需备案。
Q3:第三方认证与PMDA审批区别?
A:第三方认证适用于已有标准产品;PMDA审批适用于高风险及创新产品。
Q4:外国企业能直接申请吗?
A:不能,必须指定日本MAH。
Q5:MDSAP日本认可吗?
A:认可,可替代部分现场审核。
结语
日本市场准入门槛高,但回报同样丰厚,对于中国医疗器械企业而言:
谁能更早理解日本法规逻辑,谁就更容易抢占日本市场先机。
如果您正在布局日本市场,欢迎联系我们获取:
✅ 日本医疗器械分类支持
✅ JMDN代码确认
✅ MAH资源对接
✅ 日本QMS辅导
✅ PMDA注册咨询
EVERBIZ 深耕医疗器械全球准入合规多年,已帮助多家国内厂商成功登陆美国、欧盟(德国)、加拿大、日本及东南亚等主流市场。无论您的目标是严谨的德国市场、高门槛的日本市场,还是潜力巨大的东南亚市场,我们都能以实战经验为您定制专属合规方案,助您快速打通全球市场,实现产品价值的全球落地。
参考链接
MHLW官网:
https://www.mhlw.go.jp/english/
PMDA官网:
https://www.pmda.go.jp/english/index.html
PMDA标准数据库:
https://www.pmda.go.jp/english/review-services/standards/



